
Thủ tục công bố đủ điều kiện sản xuất trang thiết bị y tế
Thủ tục công bố tiêu chuẩn áp dụng đối với trang thiết bị y tế loại A
Xin giấy chứng nhận lưu hành tự do CFS các nước tham chiếu (Châu Âu, Anh, Úc) để đáp ứng yêu cầu thông tư Số: 14/2020/TT-BYT của Bộ Y tế trong đấu thầu trang thiết bị y tế
Tại sao cần giấy chứng nhận lưu hành tự do trong đấu thầu trang thiết bị y tế?
Theo Điều 4 của thông tư Số: 14/2020/TT-BYT: Quy định về phân nhóm và việc dự thầu vào các nhóm của gói thầu trang thiết bị y tế
1. Quy định về phân nhóm
Gói thầu trang thiết bị y tế có thể có một hoặc nhiều chủng loại trang thiết bị y tế và mỗi chủng loại trang thiết bị y tế có thể được phân chia theo các nhóm như sau:
a) Nhóm 1 gồm các trang thiết bị y tế đáp ứng đồng thời các tiêu chí sau đây:
-
Được ít nhất 02 nước trong số các nước tham chiếu* cấp giấy chứng nhận lưu hành tự do;
-
Sản xuất tại nước tham chiếu hoặc sản xuất tại Việt Nam.
b) Nhóm 2 gồm các trang thiết bị y tế đáp ứng đồng thời các tiêu chí sau đây:
-
Được ít nhất 02 nước tham chiếu cấp giấy chứng nhận lưu hành tự do;
-
Không sản xuất tại các nước tham chiếu hoặc sản xuất tại Việt Nam.
c) Nhóm 3 gồm các trang thiết bị y tế đáp ứng đồng thời các tiêu chí sau đây:
-
Được ít nhất 01 nước tham chiếu cấp giấy chứng nhận lưu hành tự do;
-
Sản xuất tại nước tham chiếu hoặc sản xuất tại Việt Nam.
d) Nhóm 4 gồm các trang thiết bị y tế đáp ứng đồng thời các tiêu chí sau đây:
-
Được ít nhất 01 nước tham chiếu cấp giấy chứng nhận lưu hành tự do;
-
Không sản xuất tại các nước tham chiếu hoặc sản xuất tại Việt Nam.
đ) Nhóm 5 gồm các trang thiết bị y tế đáp ứng đồng thời các tiêu chí sau đây:
-
Có số lưu hành tại Việt Nam (bao gồm cả các trường hợp được cấp giấy chứng nhận đăng ký lưu hành);
-
Sản xuất tại Việt Nam.
e) Nhóm 6 gồm các trang thiết bị y tế còn lại
*Danh sách nước tham chiếu:
1. Các nước khu vực Châu Âu:
-
Cộng hòa Áo;
-
Cộng hòa Ba Lan;
-
Cộng hòa Bồ Đào Nha;
-
Cộng hòa Bulgaria;
-
Cộng hòa Croatia;
-
Cộng hòa Estonia;
-
Cộng hòa Hungary;
-
Cộng hòa Hy Lạp;
-
Cộng hòa Ireland;
-
Cộng hòa Latvia;
-
Cộng hòa liên bang Đức;
-
Cộng hòa Litva;
-
Cộng hòa Malta;
-
Cộng hòa Phần Lan;
-
Cộng hòa Pháp;
-
Cộng hòa Romania;
-
Cộng hòa Séc;
-
Cộng hòa Síp;
-
Cộng hòa Slovakia;
-
Cộng hòa Slovenia;
-
Cộng hòa Tây Ban Nha;
-
Cộng hòa Ý;
-
Đại công quốc Luxembourg;
-
Liên bang Thụy Sỹ;
-
Vương quốc Bỉ;
-
Vương quốc Đan Mạch;
-
Vương quốc Hà Lan;
-
Vương quốc Thụy Điển.
2. Vương quốc Anh
3. Hợp chung quốc Hoa Kỳ.
4. Nhật Bản.
5. Thịnh vượng chung Úc.
6. Ca-na-da.
Theo đó, có đến 4 trong 6 nhóm của gói thầu trang thiết bị y tế đặt ra tiêu chí về giấy chứng nhận lưu hành tự do. Việc đáp ứng tiêu chí về giấy chứng nhận lưu hành tự do giúp trang thiết bị y tế được xếp vào nhóm tốt hơn, có cơ hội tham gia dự thầu vào nhiều nhóm hơn.
Ví dụ, nếu trang thiết bị y tế đáp ứng tiêu chí nhóm 1, trang thiết bị y tế đó sẽ được dự thầu vào các nhóm 1, 2, 3, 4, 5, 6. Tương tự với các nhóm còn lại. Nếu trang thiết bị y tế đáp ứng tiêu chí nhóm 5 thì chỉ được dự thầu vào các nhóm 5, 6. Nếu trang thiết bị y tế không đáp ứng tiêu chí tại nhóm 1, 2, 3, 4, 5 thì chỉ có thể dự thầu vào nhóm 6.
Nói tóm tại, giấy chứng nhận lưu hành tự do CFS mang đến lợi thế lớn cho nhà thầu trong đấu thầu trang thiết bị y tế. Ngoài ra, nó còn mang đến nhiều lợi ích khác cho doanh nghiệp trong quá trình kinh doanh trang thiết bị y tế.
Dịch vụ của CFOOD
Đăng ký cấp chứng nhận CFS có thể là một quá trình khó khăn, mất thời gian với các doanh nghiệp khi không có nhiều thông tin, tài liệu hướng dẫn chi tiết có thể tìm thấy. Lựa chọn dịch vụ của CFOOD, doanh nghiệp sẽ nhận được những lợi ích:
-
Có sự đồng hành của CFOOD - tổ chức chứng nhận giàu kinh nghiệm trong quá trình chuẩn bị hồ sơ, xin giấy chứng nhận lưu hành tự do CFS.
-
Được tư vấn, hướng dẫn chi tiết về thủ tục, giấy tờ. Đảm bảo chuẩn bị đầy đủ theo đúng quy định, tăng tỷ lệ xin giấy CFS thành công. Từ đó, tiết kiệm thời gian, công sức và cả chi phí của doanh nghiệp. Không làm ảnh hưởng, chậm chễ thời gian vào thầu, kinh doanh của doanh nghiệp.
-
Chi phí dịch vụ hợp lý: Với quy trình làm việc tối ưu, đội ngũ giàu kinh nghiệm, CFOOD cung cấp đến khách hàng dịch vụ tối ưu với chi phí hợp lý nhất.
CFOOD cung cấp dịch vụ hỗ trợ doanh nghiệp được cấp giấy chứng nhận lưu hành tự do CFS của Châu Âu – EU, Anh, Úc đáp ứng yêu cầu thông tư Số: 14/2020/TT-BYT của Bộ Y tế trong đấu thầu trang thiết bị y tế


.jpg)
Thủ tục công bố tiêu chuẩn áp dụng đối với trang thiết bị y tế loại A

FDA LÀ GÌ? TỔNG QUAN VỀ ĐĂNG KÝ FDA
FDA viết tắt bời cụm từ Food and Drug Adminitration- Cục quản lý Thực Phẩm và Dược phẩm Hoa Kỳ. Được thành lập từ năm 1906 tại Maryland và là cơ quan quản lý thực phẩm và dược phẩm của Hoa kỳ thuộc Bộ Y tế và Dịch vụ Nhân sinh Hoa kỳ.
HOẠT ĐỘNG CHÍNH CỦA FDA:
FDA có nhiệm vụ chịu trách nhiệm chính cho việc bảo vệ và thức đấy sức khỏe cộng đồng, ban hành và giám sát về vệ sinh an toàn thực phẩm và dược phẩm tại quốc gia mình.
FDA quy định chất lượng về:
1. Thực phẩm
2. Thực phẩm chức năng
3. Thuốc lá
4. Thiết bị y tế
5. Dược phẩm
....
GIẤY CHỨNG NHẬN FDA CÓ QUAN TRỌNG KHÔNG?
Để được xuất hàng vào mỹ là thực phẩm hay dược phẩm...., ngoài rất nhiều giấy tờ trong đó có FDA của Mỹ, nếu không có giấy tờ này hàng sẽ bị trả lại tại Hải quan Mỹ.
KHÁCH HÀNG CẦN PHẢI CUNG CẤP GÌ KHI ĐĂNG KÝ FDA?
- Các chứng nhận chất lượng ISO của cơ sở sản xuất
- Đăng ký kinh doanh
- Thông tin liên hệ
- Và một số thông tin cần tùy trường hợp
DỊCH VỤ C FOOD:
- Uy tín và chuyên nghiệp
- Đưa ra giải pháp tối ưu cho khách hàng
- Giá cả phù hợp
- Bảo mật nội dung công việc
Trong thời gian 3 ngày, kể từ khi doanh nghiệp cung cấp đủ đăng ký kinh doanh và nhãn sản phẩm thì sẽ được cấp Registration No để tiến hành xuất hàng và thông tin công ty sẽ được update vào thứ 2 đầu tuần trên Web của FDA.
FDA 510k?
FDA 510k có tên đầy đủ là FDA's Premarket Notification 510 (k) - Thông báo trước khi ra thị trường. Lý do người ta gọi 510 (k) và Premarket Notification thay thế cho nhau là vì Thông báo trước khi ra thị trường (PMN) đề cập đến mục 510 (k) của Đạo luật Thực phẩm, Dược phẩm và Mỹ phẩm Liên bang. Đây là bản đệ trình được gửi tới FDA để chứng minh rằng thiết bị y tế được tiếp thị là an toàn và hiệu quả, về cơ bản là nó tương đương với thiết bị khác được tiếp thị hợp pháp trên thị trường. Điều này có nghĩa là, cơ sở gửi đơn phải so sánh thiết bị y tế của họ với một hoặc nhiều thiết bị y tế tương tự đã được lưu hành hợp pháp ngoài thị trường, hỗ trợ các tuyên bố về tính tương đương đáng kể của họ.
Tương đương đáng kể ở (nội dung trên có đề cập đến tương đương đáng kể. Vậy, tương đương đáng kể là gì ? Tương đương đáng kể là sự chứng minh rằng thiết bị mới của doanh nghiệp, so với thiết bị đã được xác định (predicate device) có cùng mục đích sử dụng và cùng đặc điểm công nghệ hoặc có cùng mục đích sử dụng và sự khác biệt về đặc tính công nghệ nhưng không đặt ra các câu hỏi khác nhau về tính an toàn và hiệu quả Thông tin này được gửi cho FDA chứng minh rằng thiết bị an toàn và hiệu quả như thiết bị predicate.
Ai được yêu cầu gửi FDA 510k?
FDA không quy định đối tượng nào phải nộp 510 (k), họ quy định những hành động cần đệ trình 510k, ví dụ như giới thiệu, marketing thiết bị y tế tại thị trường Hoa Kỳ.
- Những hành động phải gửi FDA 510k
1. Bất kỳ ai muốn bán thiết bị y tế ở Hoa Kỳ đều phải gửi 510 (k) ít nhất 90 ngày trước khi đưa thiết bị ra bán, trừ khi nó được tiếp thị trước ngày ngày 28 tháng 5 năm 1976.
2. Có một sự thay đổi hoặc sửa đổi đối với một thiết bị được tiếp thị hợp pháp và sự thay đổi đó có thể ảnh hưởng đáng kể đến sự an toàn hoặc hiệu quả của thiết bị đó.
- Những đối tượng cần gửi FDA 510k
1. Các nhà sản xuất trong nước giới thiệu một thiết bị đến thị trường Hoa Kỳ. Lưu ý là chỉ những nhà sản xuất hoàn thiện mới được yêu cầu gửi thông báo trước khi ra thị trường . Các nhà sản xuất linh kiện không bắt buộc làm điều này, trừ khi họ tiếp thị bán linh kiện như một sản phẩm thay thế.
2. Các nhà phát triển đặc điểm kỹ thuật giới thiệu một thiết bị đến thị trường Hoa Kỳ;
3. Người đóng gói lại hoặc người gắn nhãn lại thực hiện các thay đổi về nhân hoặc có hoạt động ảnh hưởng đáng kể đến thiết bị y tế.
4. Các nhà sản xuất / xuất khẩu nước ngoài hoặc đại diện Hoa Kỳ của các nhà sản xuất / xuất khẩu nước ngoài giới thiệu thiết bị vào thị trường Hoa Kỳ.
- Trường hợp không bắt buộc phải có chứng chỉ FDA 510k
1. Bán các thiết bị chưa hoàn thiện cho một công ty khác để xử lý thêm hoặc bán các thành phần được sử dụng trong việc lắp ráp thiết bị của các công ty khác . ( Không bán cho người dùng cuối).
2. Thiết bị y tế không được tiếp thị hoặc phân phối thương mại.
3. Phân phối thiết bị sản xuất trong nước của công ty khác.
4. Đóng gói lại hoặc gắn nhãn lại và nhãn hoặc tình trạng hiện có của thiết bị không bị thay đổi đáng kể.
5. Thiết bị đã được phân phối thương mại một cách hợp pháp trước ngày 28 tháng 5 năm 1976 và không bị thay đổi hoặc chỉnh sửa đáng kể về thiết kế, thành phần, phương pháp sản xuất hoặc mục đích sử dụng.
6. Thiết bị được miễn trừ 510 (k) theo quy định (21 CFR 862-892)
510 (k) Quy trình thông quan
510 (k) là tệp chứa đầy đủ thông tin về một thiết bị để chứng minh rằng thiết bị y tế ít nhất là an toàn và hiệu quả, tương tự như thiết bị được tiếp thị hợp pháp không phải tuân theo PMA.
Các tổ chức dự định tung ra các Thiết bị Y tế Loại I, Loại II và Loại III ở Hoa Kỳ nhằm mục đích sử dụng cho con người phải gửi US FDA 510 (k) nếu không yêu cầu Phê duyệt trước khi đưa ra thị trường (PMA).
Hầu hết các thiết bị loại I được miễn yêu cầu 510 (k). Trong quá trình xem xét tệp 510 (k), nếu FDA nhận thấy thiết bị là Tương đương Cơ bản (SE), FDA sẽ đưa ra thông quan bằng số 'k'.
Giải pháp cho FDA 510k
Chúng tôi đưa ra lời khuyên của chuyên gia, Phân tích thiết bị dự đoán, Mã sản phẩm cùng với nhận dạng số quy định, chuẩn bị 510 (k), Xác định yêu cầu và tiêu chuẩn thử nghiệm, dịch vụ Đại lý Hoa Kỳ, Q-Submission, phản hồi đánh giá của FDA và cập nhật được đề cập trong các dịch vụ của chúng tôi.
Hãy liên hệ với chúng tôi để nhận báo giá và các dịch vụ chi tiết cho các sản phẩm:
- FDA CODE : LZA (Nitrile Examination Gloves)
- FDA CODE : LYY (Latex Examination Gloves)
- FDA CODE – FXX / OUK
- FDA CODE – FMF (Piston Syringe)
- FDA CODE – FMI (Disposable Hypodermic Syringe)
- FDA CODE – FMI + FMF (Disposable Hypodermic Syringe with Piston)
- FDA CODE : FYA (Surgical Gown)
- FDA CODE : OEA (Isolation Gown)
Chứng nhận CE Marking được xem như “hộ chiếu thương mại” của các quốc gia khu vực Châu Âu. Với nhu cầu hội nhập toàn cầu như hiện nay, xuất nhập khẩu hàng hóa đã quá đỗi quen thuộc. Một sản phẩm được dán dấu CE lên trên, sẽ được lưu thông tự do trên thị trường Châu Âu.
Tìm hiểu về CE Marking
Conformité Européenne là tên tiếng Pháp đầy đủ của CE, dịch ra tiếng Việt có nghĩa là tiêu chuẩn Châu Âu. CE Marking là tên chính thức của CE.
Một sản phẩm nếu đạt được chứng nhận CE Marking đồng nghĩa với việc nó có thể lưu thông tự do trong thị trường Châu Âu, được pháp luật của Liên minh Châu âu EU công nhận.
Một điểm đáng chú ý nữa là dấu CE. Đây như là con dấu chứng nhận một sản phẩm đạt tiêu chuẩn CE. Việc dán dấu này không hề đơn giản, được EU quy định rất nghiêm ngặt. Mỗi loại sản phẩm sẽ có cách dán tương ứng khác nhau, và những quy định giống nhau như:
- Tỉ lệ của biểu tượng dấu CE phải luôn được giữ nguyên, chỉ có kích thước tăng lên hoặc giảm xuống (không được dưới 5mm).
- Việc dán dấu CE phải thật tỉ mỉ, đảm bảo biểu tượng CE phải được đặt thẳng đứng, và không bị che bởi các logo khác.
Dấu CE là một biểu tượng phải được gắn vào nhiều sản phẩm trước khi chúng có thể được bán trên thị trường châu Âu. Dấu hiệu chỉ ra rằng một sản phẩm:
- Hoàn thành các yêu cầu của chỉ thị sản phẩm châu Âu có liên quan
- Đáp ứng tất cả các yêu cầu của các tiêu chuẩn an toàn và hiệu suất hài hòa được công nhận của Châu Âu
- Phù hợp với mục đích của nó và sẽ không gây nguy hiểm đến tính mạng hoặc tài sản
Mỗi dấu CE sẽ đại diện cho nhà sản xuất, từ đó nhà sản xuất phải có trách nhiệm pháp lý với sản phẩm đó theo quy định hiện hành. Một nguyên tắc phải luôn nhớ là khi dán dấu CE, bạn không được đính kém với bất cứ một sản phẩm nào khác, phải tuân theo quy định riêng của EU.
Việc gắn Dấu CE vào sản phẩm được coi là một phương tiện để chứng nhận cho các cơ quan có thẩm quyền trong các quốc gia thành viên EU rằng sản phẩm đáp ứng tất cả các yêu cầu phù hợp của EU.
Có một yêu cầu của EU rằng các sản phẩm không phù hợp với các quy định của các chỉ thị không được phép lưu hành trong lãnh thổ của các quốc gia thành viên; hành động thích hợp nên được thực hiện để loại bỏ các sản phẩm này khỏi bán và sử dụng trong trạng thái cụ thể. Một ví dụ là việc nhập khẩu đồ chơi gần đây từ Trung Quốc sang Anh, khi được kiểm tra, được phát hiện có chứa chất độc độc cao gây nguy hiểm đến tính mạng.
Nhà nhập khẩu và / hoặc nhà sản xuất phải thực hiện các bước để tuân thủ các quy định an toàn, xuất trình các hồ sơ phù hợp và quyết định các quy trình cần thiết để duy trì sản xuất tuân thủ các chỉ thị. Dấu CE phải được dán để chứng minh sự phù hợp với các quy định của chỉ thị.
Các chỉ thị cụ thể có các mục tiêu an toàn toàn diện, nhưng chúng khiến nhà sản xuất đưa ra quyết định về cách thức đạt được những điều này.
Khi có nhiều chỉ thị CE Mark liên quan đến sản phẩm và giai đoạn chuyển tiếp cho phép nhà sản xuất lựa chọn áp dụng, việc đánh dấu chỉ cho thấy sự phù hợp với các chỉ thị được nhà sản xuất áp dụng. Trong trường hợp này, các chỉ thị đã được áp dụng phải được xác định trong các tài liệu hoặc thông báo kèm theo sản phẩm. Trong trường hợp nhà sản xuất không liệt kê những chỉ thị đã được áp dụng, chính quyền sẽ cho rằng một tuyên bố về sự phù hợp có sẵn cho tất cả các chỉ thị hiện hành
Khi nào đánh dấu CE là bắt buộc?
Đánh dấu CE chỉ bắt buộc đối với các sản phẩm có thông số kỹ thuật của EU tồn tại và yêu cầu gắn dấu CE.Một số sản phẩm phải tuân theo một số yêu cầu của EU cùng một lúc. Bạn phải đảm bảo rằng sản phẩm của bạn tuân thủ tất cả các yêu cầu có liên quan trước khi gắn dấu CE vào nó. Nghiêm cấm gắn dấu CE vào các sản phẩm mà thông số kỹ thuật của EU không tồn tại hoặc không yêu cầu gắn dấu CE.
Đối tượng áp dụng tiêu chuẩn CE
Tiêu chuẩn CE ban đầu là được ban hành cho các quốc gia thành viên của Liên Minh Châu Âu. Tiếp đó sử dụng cho thành viên Hiệp hội Thương mại Tự do EFTA. Sau đó, với nhu cầu trao đổi hàng hóa tăng mạnh, dấu CE được dùng phổ biến hơn.Các mặt hàng từ các quốc gia khác khi muốn xuất sang thị trường Châu Âu đều phải có chứng chỉ CE Marking, ví dụ như:
Dấu CE áp dụng cho các sản phẩm, từ thiết bị điện đến đồ chơi và từ chất nổ dân dụng đến thiết bị y tế.
Danh sách đầy đủ của các loại sản phẩm dưới đây:
- thiết bị y tế cấy ghép hoạt động
- thiết bị đốt nhiên liệu khí
- lắp đặt cáp treo được thiết kế để chở người
- thiết kế sinh thái các sản phẩm liên quan đến năng lượng
- tương thích điện từ
- thiết bị và hệ thống bảo vệ dự định sử dụng trong môi trường có khả năng gây nổ
- chất nổ dùng trong dân dụng
- nồi hơi nước nóng
- tủ lạnh gia đình và tủ đông
- thiết bị y tế chẩn đoán in vitro
- thang máy
- điện áp thấp
- máy móc
- dụng cụ đo lường
- các thiết bị y tế
- tiếng ồn trong môi trường
- dụng cụ cân không tự động
- thiết bị bảo vệ cá nhân
- thiết bị áp suất
- pháo hoa
- thiết bị đầu cuối đài phát thanh và viễn thông
- nghề giải trí
- an toàn của đồ chơi
- bình áp lực đơn giản
.jpg)
Dấu CE không bắt buộc đối với các mục, ví dụ:
- hóa chất
- dược phẩm
- mỹ phẩm và thực phẩm
Những lợi ích khi tham gia chứng nhận CE Marking
Như đã giới thiệu từ đầu, chứng nhận CE Marking giống như lá phiếu thông hành, từ đó giúp hàng hóa của bạn thuận tiện lưu thông. Khi có chứng chỉ CE Marking, bạn sẽ nhận được rất nhiều lợi ích và cơ hội phát triển từ nó.- Một sản phẩm được dán dấu CE, đồng nghĩa chất lượng của nó cực tốt, đạt chuẩn Châu Âu. Từ đó, việc mở rộng thị trường trong và ngoài nước là rất dễ dàng. Khi hàng được xuất khẩu ra nước bạn, giá trị sản phẩm sẽ tăng lên rất nhiều.
- Giúp bạn nâng cao giá trị thương hiệu, đẩy mạnh tính cạnh tranh của sản phẩm. Việc kinh doanh của bạn sẽ phát triển với lợi nhuận cao hơn.
- Nói đến chất lượng đạt chuẩn Châu Âu thì khách hàng nào cũng mong muốn sở hữu. Sản phẩm chất lượng tốt sẽ giành được lòng tin và sự yêu mến của khách hàng. Đây là chiến lược kinh doanh hàng đầu của mọi thương hiệu.
- Dấu CE đảm bảo sản phẩm của bạn có thể vào Liên minh châu Âu và cho phép di chuyển tự do trên gần 30 quốc gia tạo nên Khu vực kinh tế châu Âu, cho phép bạn tiếp cận trực tiếp với hơn 500 triệu người tiêu dùng. Nếu một sản phẩm nên hiển thị dấu CE không được tìm thấy, nhà sản xuất hoặc nhà phân phối có thể bị phạt và phải đối mặt với việc thu hồi sản phẩm đắt tiền – vì vậy việc tuân thủ là rất cần thiết.
Cách đặt dấu CE trên sản phẩm
Trước khi bạn đặt dấu CE trên sản phẩm, bạn cần thiết lập Chỉ thị tiếp cận mới của EU áp dụng cho sản phẩm của mình. Bạn không được đính kèm dấu CE vào sản phẩm ngoài phạm vi chỉ thị.Quá trình bạn tuân theo phụ thuộc vào các chỉ thị áp dụng cho sản phẩm của bạn.
1. Xác định (các) chỉ thị và các tiêu chuẩn hài hòa áp dụng cho sản phẩm
Có hơn 20 chỉ thị đặt ra các loại sản phẩm yêu cầu đánh dấu CE. Các yêu cầu thiết yếu mà các sản phẩm phải đáp ứng, ví dụ như an toàn, được tạo ra ở cấp EU và được quy định trong các điều khoản chung trong các chỉ thị này. Các tiêu chuẩn châu Âu hài hòa được ban hành với tham chiếu đến các chỉ thị được áp dụng và thể hiện các yêu cầu an toàn thiết yếu trong các điều khoản kỹ thuật chi tiết.2. Kiểm tra các yêu cầu cụ thể của sản phẩm
Tùy thuộc vào bạn để đảm bảo rằng sản phẩm của bạn tuân thủ các yêu cầu thiết yếu của pháp luật EU có liên quan . Việc sử dụng các tiêu chuẩn hài hòa vẫn là tự nguyện. Bạn có thể quyết định chọn những cách khác để đáp ứng các yêu cầu thiết yếu này. Nếu bạn không tuân theo các yêu cầu an toàn của tiêu chuẩn vì nó được viết, bạn sẽ cần chứng minh rằng sản phẩm của bạn an toàn, bằng cách xuất trình các tài liệu liên quan.3. Xác định xem có cần phải đánh giá sự phù hợp độc lập từ Cơ quan thông báo không
Mỗi chỉ thị bao gồm sản phẩm của bạn chỉ định liệu bên thứ ba được ủy quyền (Cơ quan thông báo) có phải tham gia vào quy trình đánh giá sự phù hợp cần thiết để đánh dấu CE hay không. Điều này không bắt buộc đối với tất cả các sản phẩm, vì vậy điều quan trọng là phải kiểm tra xem có cần phải có sự tham gia của Cơ quan Thông báo hay không. Các cơ quan này được ủy quyền bởi các cơ quan quốc gia và chính thức ‘thông báo’ cho Ủy ban Châu Âu và được liệt kê trên cơ sở dữ liệu của NANDO (Tổ chức được thông báo và chỉ định tiếp cận mới) .4. Kiểm tra sản phẩm và kiểm tra sự phù hợp
Nếu bạn sản xuất một sản phẩm, bạn có trách nhiệm kiểm tra sản phẩm và kiểm tra sự phù hợp của nó với luật pháp EU (quy trình đánh giá sự phù hợp). Một phần của thủ tục là, theo nguyên tắc chung, đánh giá rủi ro. Bằng cách áp dụng các tiêu chuẩn châu Âu hài hòa có liên quan, bạn sẽ có thể thực hiện các yêu cầu lập pháp cần thiết của các chỉ thị.5. Vẽ và giữ sẵn các tài liệu kỹ thuật cần thiết
Nếu bạn sản xuất một sản phẩm, bạn cần thiết lập các tài liệu kỹ thuật theo yêu cầu của (các) chỉ thị để đánh giá sự phù hợp của sản phẩm với các yêu cầu liên quan và để đánh giá rủi ro. Bạn phải có thể xuất trình tài liệu kỹ thuật và EC DoC cho các cơ quan quốc gia có liên quan, nếu được yêu cầu.6. Đặt dấu CE trên sản phẩm của bạn và Tuyên bố về sự phù hợp của EC
Dấu CE phải được đặt trên sản phẩm bởi nhà sản xuất hoặc bởi đại diện ủy quyền của anh ta trong EEA hoặc Thổ Nhĩ Kỳ. Nó phải được đặt theo định dạng hợp pháp của nó cho sản phẩm hoặc bảng dữ liệu của nó. Nó phải được nhìn thấy, dễ đọc và không thể loại bỏ. Nếu một Thông báo có liên quan đến giai đoạn kiểm soát sản xuất, số nhận dạng của nó cũng phải được hiển thị. Nhà sản xuất có trách nhiệm lập và ký một ‘ EC DoC ‘ chứng minh rằng sản phẩm đáp ứng các yêu cầu. Thế là xong, sản phẩm được đánh dấu CE của bạn đã sẵn sàng cho thị trường.Quy định dán nhãn CE lên sản phẩm
Một số quy định chung như sau :- Kích thước của biểu tượng dấu " CE " khi tăng hay giảm thì tỷ lệ vẫn phải được giữ nguyên
- Dấu " CE " được đặt theo chiều thẳng đứng và kích thước không được nhỏ hơn 5mm Dấu " CE " phải đặt ở vị trí không bị các logo khác che khuất Dấu CE được in trên sản phẩm đồng nghĩa với việc công bố sản phẩm thỏa mãn những yêu cầu của châu Âu , và được chấp nhận tại hầu hết các nước trên thế giới Chứng nhận CE là bắt buộc đối với hàng hóa và được coi như hộ chiếu thương mại vào thị trường Châu Âu ( và cả một số nước khác ngoài Châu Âu như Mỹ , Malaysia, Úc, Iran ... ). Khi tổ chức đánh giá kiểm tra sản phẩm phù hợp với các yêu cầu của các tiêu chuẩn CE thì nhà sản xuất sẽ được họ cấp giấy chứng nhận CE, lúc đó nhà nhà xuất có thể gắn dấu CE trên sản phẩm của mình.
Hãy liên hệ với chúng tôi để nhận báo giá và các dịch vụ chi tiết.



Link tra cứu: https://uasl.uk.com/certifiedorganization/
Ngoài ra CFOOD còn cung cấp dịch vụ làm CE SATTRA 2777 cho Thiết bị bảo hộ cá nhân theo Regulation (EU) 2016/425 Personal protective equipment
Thiết bị bảo vệ cá nhân (PPE) là các sản phẩm mà người dùng có thể đeo hoặc cầm để được bảo vệ khỏi các rủi ro tại nơi làm việc, ở nhà hoặc trong khi tham gia các hoạt động giải trí. Số liệu thống kê về tai nạn lao động chết người và lớn nhấn mạnh tầm quan trọng của việc bảo vệ và phòng ngừa, trong đó phương tiện bảo vệ cá nhân đóng một vai trò quan trọng.
Luật pháp EU và PPE
Quy định (EU) 2016/425 ngày 9 tháng 3 năm 2016 về thiết bị bảo vệ cá nhân ( quy định PPE ) bao gồm việc thiết kế, sản xuất và tiếp thị thiết bị bảo vệ cá nhân . Nó xác định các nghĩa vụ pháp lý để đảm bảo rằng PPE trên thị trường nội bộ EU cung cấp mức độ bảo vệ cao nhất trước các rủi ro. Các dấu CE gắn liền với PPE cung cấp bằng chứng về sự phù hợp của sản phẩm với quy định của EU áp dụng.
Do pháp luật dựa trên 'cách tiếp cận mới' phù hợp với chính sách khung lập pháp mới , các nhà sản xuất hoặc đại diện được ủy quyền của họ tại EU phải tuân thủ các yêu cầu thiết yếu về sức khỏe và an toàn của quy định PPE , trực tiếp hoặc bằng cách sử dụng các tiêu chuẩn hài hòa của Châu Âu. Sau này giả định về sự phù hợp với các yêu cầu pháp lý.
Các hướng dẫn về quy định PPE (ấn bản lần 1 - tháng 4 năm 2018) (6 MB) nhằm mục đích tạo điều kiện cho sự hiểu biết chung và thực hiện các quy định về PPE .
Quy định PPE được áp dụng từ ngày 21 tháng 4 năm 2018 , thay thế Chỉ thị 89/686 / EEC trước đó.
.jpg)
LƯU HÀNH SẢN PHẨM THIẾT BỊ Y TẾ TẠI THỊ TRƯỜNG CHÂU ÂU

Đại diện Châu Âu (EC REP) là gì? Tầm quan trọng của việc đăng ký đại diện Châu Âu
Điều khoản 14 (2) của chỉ thị về trang thiết bị y tế nêu rõ: “Trường hợp nhà sản xuất đưa thiết bị ra thị trường dưới tên của chính mình mà không có địa điểm kinh doanh đã đăng ký tại Quốc gia Thành viên ở Liên Minh Châu Âu, thì nhà sản xuất đó cần chỉ định một đại diện được ủy quyền tại Liên minh Châu Âu.”
Tên và địa chỉ của Người đại diện được ủy quyền phải được ghi trên nhãn, bao bì bên ngoài hoặc hướng dẫn sử dụng (IFU). Công ty bạn cũng phải sử dụng ký hiệu Đại diện EC như được chỉ định trong EN ISO 15223-1: 2016. Theo MDR (EU) mới 2017/745 Đại diện được ủy quyền (Đại diện EC) có trách nhiệm lớn hơn và chịu nhiều rủi ro và trách nhiệm pháp lý hơn đáng kể, do đó, bạn có thể mong đợi người đại diện xem xét tài liệu của bạn kỹ lưỡng hơn.
Căn cứ trên nhu cầu của khách hàng, chúng tôi xin gửi Quý khách các dịch vụ tư vấn để có thể lưu hành sản phẩm tại thị trường Châu Âu cho từng sản phẩm, cụ thể như sau:
Dịch vụ đại diện ủy quyền Châu Âu
Đăng ký số lưu hành cho sản phẩm
CFS lưu hành tự do
Hãy liên hệ với chúng tôi để nhận báo giá và các dịch vụ chi tiết cho các sản phẩm.


Đăng ký công bố tiêu chuẩn áp dụng đối với thiết bị y tế loại A, B

Xin giấy chứng nhận lưu hành tự do CFS để đáp ứng yêu cầu thông tư Số: 14/2020/TT-BYT của Bộ Y tế trong đấu thầu trang thiết bị y tế
THỦ TỤC THÀNH LẬP CÔNG TY SẢN XUẤT GĂNG TAY Y TẾ

Hướng dẫn giấy phép lưu hành tự do Găng Tay y tế

SỐ DUNS LÀ GÌ? BAO GỒM NHỮNG GÌ VÀ LỢI ÍCH CỦA DUNS NHƯ THẾ NÀO?

TƯ VẤN ĐIỀU KIỆN SẢN XUẤT KHẨU TRANG Y TẾ

CẤP CHỨNG NHẬN CE MARKING CHO THIẾT BỊ Y TẾ THEO CHỈ THỊ 93/42/EEC
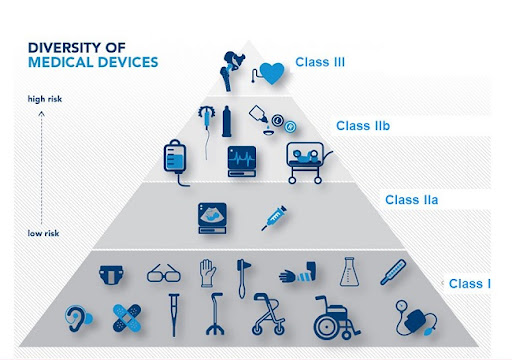
PHÂN LOẠI THIẾT BỊ Y TẾ VÀ ĐỊNH TUYẾN CỦA CHÚNG ĐỂ ĐÁNH DẤU CE






























































Địa chỉ công ty: Số 52 ngõ 45 đường Võ Chí Công, phường Nghĩa Đô, thành phố Hà Nội, Việt Nam
Địa chỉ VP: Số 34 TT02, KĐT HD Mon, đường Hàm Nghi, phường Từ Liêm, thành phố Hà Nội
Điện thoại: 0904.699.600
Email : info@cfood.com.vn
 Hôm nay :
1.464
Hôm nay :
1.464
![]() Đang online :
6
Đang online :
6
![]() Tuần này :
3.004
Tuần này :
3.004
![]() Tháng này :
10.835
Tháng này :
10.835
![]() Tổng truy cập :
30.197
Tổng truy cập :
30.197